Source code for the pusher proteins model in LAMMPS.
Copy the following files into LAMMPS /src directory and compile as usual. Requies the ASPHERE and MOLECULE packages to be installed.
Has been tested on 30Sept-13 and 10Aug-15 versions of LAMMPS.
pair_push.cpp
pair_push.h
Updated code work with newer lammps versions (tested on lammps 12Dec18 version)
pair_push.cpp
pair_push.h
The system must be set up with a torsionally rigid polymer (though the torsion enegry can be set to zero) as described here.
The interaction must be between an ellipsoid type atom, and a non-ellipsoid atom. LAMMPS will throw an error if this is not the case.
The equation for the interaction is:
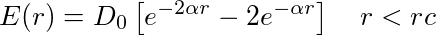
and an extra force f is added in the direction axis of the z-axis of the ellipsoid atom.
In LAMMPS an example input would be
pair_style hybrid lj/cut 1.12246152962189 push 1.0
pair_modify shift yes
pair_coeff 1 1 lj/cut 1.0 1.0 1.12246152962189
pair_coeff 1 2 push 30.0 2.0 1.0 1.0
pair_coeff 2 2 lj/cut 1.0 1.0 1.12246152962189
where type 1 are ellipsoid atoms in a polymer, and type 2 are the pusher proteins and are non-ellipsoid atoms. The order of the parameters is:
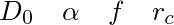
Here is an example input file and script:
in.DNA
lammps.input